GSEApy
June 24, 2026 · View on GitHub
GSEApy
GSEApy: Gene Set Enrichment Analysis in Python.
.. image:: https://badge.fury.io/py/gseapy.svg :target: https://badge.fury.io/py/gseapy
{kind=link}
.. image:: https://img.shields.io/conda/vn/bioconda/GSEApy.svg?style=plastic :target: http://bioconda.github.io
{kind=link}
.. image:: https://anaconda.org/bioconda/gseapy/badges/downloads.svg
:target: https://anaconda.org/bioconda/gseapy
{kind=link}
.. image:: https://github.com/zqfang/GSEApy/workflows/GSEApy/badge.svg?branch=master :target: https://github.com/zqfang/GSEApy/actions :alt: Action Status
{kind=link}
.. image:: http://readthedocs.org/projects/gseapy/badge/?version=master :target: http://gseapy.readthedocs.io/en/master/?badge=master :alt: Documentation Status
.. image:: https://img.shields.io/badge/license-MIT-blue.svg :target: https://img.shields.io/badge/license-MIT-blue.svg
{kind=link}
.. image:: https://img.shields.io/pypi/pyversions/gseapy.svg :alt: PyPI - Python Version
{kind=link}
Release notes : https://github.com/zqfang/GSEApy/releases
Tutorial for general usage <https://gseapy.readthedocs.io/en/latest/gseapy_example.html>_
- A faithful Rust port of fgsea's multilevel algorithm that resolves p-values far below
1/permutation_num.
Citation
::
Zhuoqing Fang, Xinyuan Liu, Gary Peltz, GSEApy: a comprehensive package for performing gene set enrichment analysis in Python,
Bioinformatics, 2022;, btac757, https://doi.org/10.1093/bioinformatics/btac757
GSEApy is a Python/Rust implementation for GSEA and wrapper for Enrichr.
GSEApy can be used for RNA-seq, ChIP-seq, Microarray data. It can be used for convenient GO enrichment and to produce publication quality figures in python.
GSEApy has 7 sub-commands available: gsea, prerank, ssgsea, gsva, replot enrichr, biomart.
:gsea: The gsea module produces GSEA <http://www.broadinstitute.org/cancer/software/gsea/wiki/index.php/Main_Page>_ results. The input requries a txt file(FPKM, Expected Counts, TPM, et.al), a cls file, and gene_sets file in gmt format.
:prerank: The prerank module produces Prerank tool results. The input expects a pre-ranked gene list dataset with correlation values, provided in .rnk format, and gene_sets file in gmt format. prerank module is an API to GSEA pre-rank tools. It also supports the fgsea multilevel p-value method (a faithful Rust port of fgsea <https://github.com/ctlab/fgsea>) via method='multilevel', which resolves p-values far below 1/permutation_num.
:ssgsea: The ssgsea module performs single sample GSEA(ssGSEA) analysis. The input expects a pd.Series (indexed by gene name), or a pd.DataFrame (include GCT file) with expression values and a GMT file. For multiple sample input, ssGSEA reconigzes gct format, too. ssGSEA enrichment score for the gene set is described by D. Barbie et al 2009 <http://www.nature.com/nature/journal/v462/n7269/abs/nature08460.html>.
:gsva: The gsva module performs GSVA <https://github.com/rcastelo/GSVA>_ method by Hänzelmann et al <https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-7>_. The input is same to ssgsea.
:replot: The replot module reproduce GSEA desktop version results. The only input for GSEApy is the location to GSEA Desktop output results.
:enrichr: The enrichr module enable you perform gene set enrichment analysis using Enrichr API. Enrichr is open source and freely available online at: http://amp.pharm.mssm.edu/Enrichr . It runs very fast.
:biomart: The biomart module helps you convert gene ids using BioMart API.
Please use 'gseapy COMMAND -h' to see the detail description for each option of each module.
The full GSEA is far too extensive to describe here; see
GSEA <http://www.broadinstitute.org/cancer/software/gsea/wiki/index.php/Main_Page>_ documentation for more information. All files' formats for GSEApy are identical to GSEA desktop version.
Why GSEApy
I would like to use Pandas to explore my data, but I did not find a convenient tool to do gene set enrichment analysis in python. So, here are my reasons:
- Ability to run inside python interactive console without having to switch to R!!!
- User friendly for both wet and dry lab users.
- Produce or reproduce publishable figures.
- Perform batch jobs easy.
- Easy to use in bash shell or your data analysis workflow, e.g. snakemake.
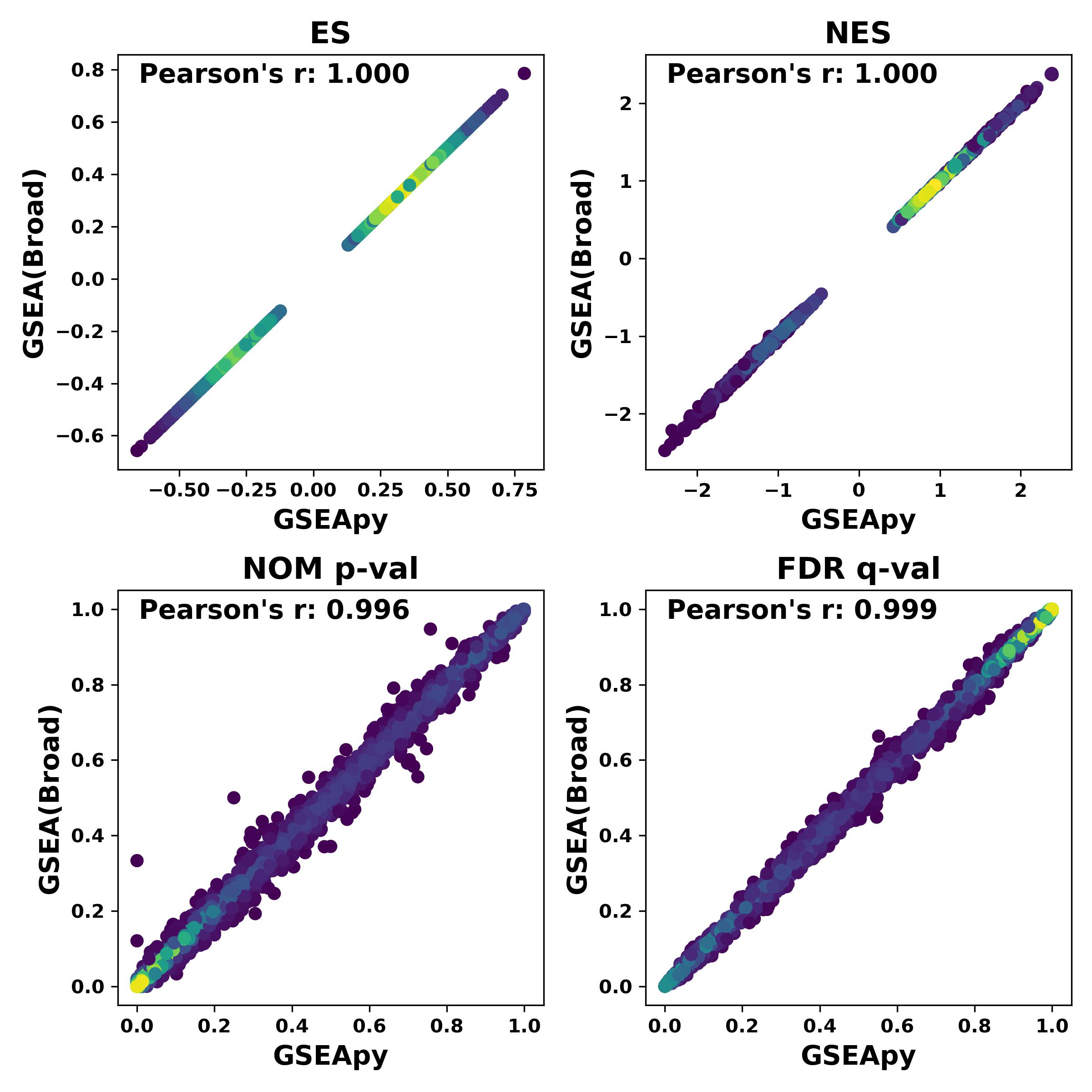
GSEApy vs GSEA(Broad) output
Using the same data for GSEAPreranked, and GSEApy reproduce similar results.
.. image:: https://raw.githubusercontent.com/zqfang/GSEApy/master/docs/Preank.py.vs.broad.jpg :width: 400
{kind=link}
See more output here: Example <http://gseapy.readthedocs.io/en/master/gseapy_example.html>_
Installation
| Install gseapy package from bioconda or pip.
.. code:: shell
if you have conda/mamba
$ conda install -c bioconda gseapy
or pip
$ pip install gseapy
or uv
$ uv add gseapy
| If pip install failed, install Rust first and build from source:
.. code:: shell
install rust toolchain
curl https://sh.rustup.rs -sSf | sh -s -- -y export PATH="HOME/.cargo/bin"
then install via pip or uv
$ pip install gseapy
or
$ uv add gseapy
Dependency
- Python 3.8+
Mandatory
* build
* Rust: For gseapy > 0.11.0, Rust compiler is needed
* setuptools-rust
* run
* Numpy >= 1.13.0
* Scipy
* Pandas
* Matplotlib
* Requests
Run GSEApy
-----------------
For command line usage:
.. code:: bash
An example to reproduce figures using replot module.
$ gseapy replot -i ./Gsea.reports -o test
An example to run GSEA using gseapy gsea module
$ gseapy gsea -d exptable.txt -c test.cls -g gene_sets.gmt -o test
An example to run Prerank using gseapy prerank module
$ gseapy prerank -r gsea_data.rnk -g gene_sets.gmt -o test
An example to run Prerank with the fgsea multilevel p-value method
$ gseapy prerank -r gsea_data.rnk -g gene_sets.gmt --method multilevel --sample-size 101 --eps 1e-50 -o test
An example to run ssGSEA using gseapy ssgsea module
$ gseapy ssgsea -d expression.txt -g gene_sets.gmt -o test
An example to run GSVA using gseapy ssgsea module
$ gseapy gsva -d expression.txt -g gene_sets.gmt -o test
An example to use enrichr api
see details for -g input -> get_library_name
$ gseapy enrichr -i gene_list.txt -g KEGG_2016 -o test
Run gseapy inside python console:
1. Prepare expression.txt, gene_sets.gmt and test.cls required by GSEA, you could do this
.. code:: python
import gseapy
# run GSEA.
gseapy.gsea(data='expression.txt', gene_sets='gene_sets.gmt', cls='test.cls', outdir='test')
# run prerank
gseapy.prerank(rnk='gsea_data.rnk', gene_sets='gene_sets.gmt', outdir='test')
# run prerank with the fgsea multilevel p-value method
gseapy.prerank(rnk='gsea_data.rnk', gene_sets='gene_sets.gmt', method='multilevel', outdir='test')
# run ssGSEA
gseapy.ssgsea(data="expression.txt", gene_sets= "gene_sets.gmt", outdir='test')
# run GSVA
gseapy.gsva(data="expression.txt", gene_sets= "gene_sets.gmt", outdir='test')
# An example to reproduce figures using replot module.
gseapy.replot(indir='./Gsea.reports', outdir='test')
2. If you prefer to use Dataframe, dict, list in interactive python console, you could do this.
see detail here: `Example <http://gseapy.readthedocs.io/en/master/gseapy_example.html>`_
.. code:: python
# assign dataframe, and use enrichr library data set 'KEGG_2016'
expression_dataframe = pd.DataFrame()
sample_name = ['A','A','A','B','B','B'] # always only two group,any names you like
# assign gene_sets parameter with enrichr library name or gmt file on your local computer.
gseapy.gsea(data=expression_dataframe, gene_sets='KEGG_2016', cls= sample_names, outdir='test')
# prerank tool
gene_ranked_dataframe = pd.DataFrame()
gseapy.prerank(rnk=gene_ranked_dataframe, gene_sets='KEGG_2016', outdir='test')
# ssGSEA
gseapy.ssgsea(data=expression_dataframe, gene_sets='KEGG_2016', outdir='test')
# gsva
gseapy.gsva(data=expression_dataframe, gene_sets='KEGG_2016', outdir='test')
3. For ``enrichr`` , you could assign a list, pd.Series, pd.DataFrame object, or a txt file (should be one gene name per row.)
.. code:: python
# assign a list object to enrichr
gl = ['SCARA3', 'LOC100044683', 'CMBL', 'CLIC6', 'IL13RA1', 'TACSTD2', 'DKKL1', 'CSF1',
'SYNPO2L', 'TINAGL1', 'PTX3', 'BGN', 'HERC1', 'EFNA1', 'CIB2', 'PMP22', 'TMEM173']
gseapy.enrichr(gene_list=gl, gene_sets='KEGG_2016', outdir='test')
# or a txt file path.
gseapy.enrichr(gene_list='gene_list.txt', gene_sets='KEGG_2016',
outdir='test', cutoff=0.05, format='png' )
GSEApy supported gene set libaries :
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
To see the full list of gseapy supported gene set libraries, please click here: `Library <http://amp.pharm.mssm.edu/Enrichr/#stats>`_
Or use ``get_library_name`` function inside python console.
.. code:: python
#see full list of latest enrichr library names, which will pass to -g parameter:
names = gseapy.get_library_name()
# show top 20 entries.
print(names[:20])
['Genome_Browser_PWMs',
'TRANSFAC_and_JASPAR_PWMs',
'ChEA_2013',
'Drug_Perturbations_from_GEO_2014',
'ENCODE_TF_ChIP-seq_2014',
'BioCarta_2013',
'Reactome_2013',
'WikiPathways_2013',
'Disease_Signatures_from_GEO_up_2014',
'KEGG_2016',
'TF-LOF_Expression_from_GEO',
'TargetScan_microRNA',
'PPI_Hub_Proteins',
'GO_Molecular_Function_2015',
'GeneSigDB',
'Chromosome_Location',
'Human_Gene_Atlas',
'Mouse_Gene_Atlas',
'GO_Cellular_Component_2015',
'GO_Biological_Process_2015',
'Human_Phenotype_Ontology',]
Dev
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
.. code:: shell
# clone and set up dev environment (requires Rust toolchain)
$ git clone https://github.com/zqfang/GSEApy.git
$ cd GSEApy
$ uv sync --extra dev
# run tests
$ uv run pytest
# lint and format
$ uv run ruff format gseapy
$ uv run ruff check gseapy
# test rust extension only
$ cargo test --features=extension-module
# build wheel + sdist locally
$ uv build
Bug Report
~~~~~~~~~~~~~~~~~~~~~~~~~~~
If you would like to report any bugs when use gseapy, don't hesitate to create an issue on github here.
To get help of GSEApy
------------------------------------
1. See `Frequently Asked Questions <https://gseapy.readthedocs.io/en/latest/faq.html>`_
2. Visit the document site at `Examples <https://gseapy.readthedocs.io/en/latest/gseapy_example.html>`_
3. The GSEApy discussion channel: `Q&A <https://github.com/zqfang/GSEApy/discussions>`_